Pharmacotherapeutic Group: Psychostimulants.
Pharmacology: Pharmacodynamics: Mechanism of action: Ritalin is a racemate consisting of a 1:1 mixture of d-methylphenidate (d-MPH) and l-methylphenidate (l-MPH).
Ritalin is a mild CNS stimulant with more prominent effects on mental than on motor activities. Its mode of action in humans is not completely understood, but its stimulant effects are thought to be due to an inhibition of dopamine and norepinephrine reuptake into presynaptic neurons and thereby increasing these neurotransmitters in the extraneuronal space.
The mechanism by which Ritalin exerts its mental and behavioral effects in children is not clearly established, nor is there conclusive evidence showing how these effects relate to the condition of the central nervous system.
The l-enantiomer is thought to be pharmacologically inactive.
The effect of treatment with 40 mg dexmethylphenidate hydrochloride, the pharmacologically active d-enantiomer of Ritalin, on QT/QTc interval was evaluated in a study in 75 healthy volunteers. The maximum mean prolongation of QTcF intervals was <5 ms, and the upper limit of the 90% confidence interval was below 10 ms for all time matched comparisons versus placebo. This was below the threshold of clinical concern and no exposure response relationship was evident.
Clinical Studies: Ritalin has been used for over 50 years in the treatment of ADHD. Its effectiveness in the treatment of ADHD is well established. In addition to improving core symptoms of ADHD, methylphenidate also improves behaviors associated with ADHD such as impaired academic performance and social function.
Studies in the published literature have shown Ritalin to significantly improve daytime sleepiness and cataplexy.
Children with ADHD: Ritalin LA was evaluated in a randomized, double-blind, placebo-controlled, parallel group clinical study in which 134 children, ages 6 to 12, with DSM-IV diagnoses of Attention Deficit Hyperactivity Disorder (ADHD) received a single morning dose of Ritalin LA in the range of 10-40 mg/day, or placebo, for up to 2 weeks. The optimal dose for each patient was determined in a dose titration phase of the study prior to randomization.
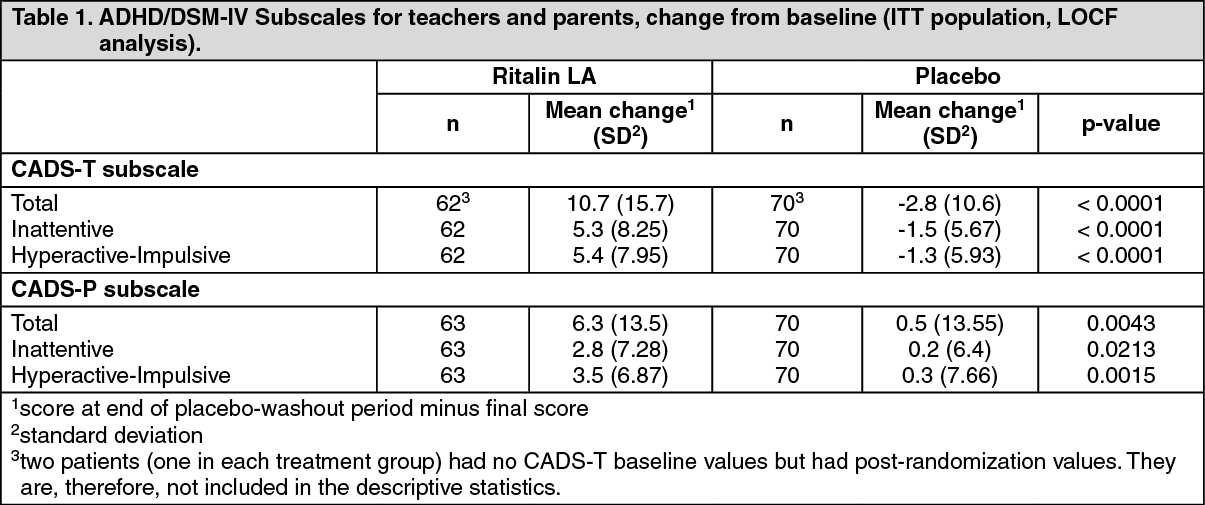
The primary efficacy variable was the change from baseline to the final rating in the ADHD/DSM-IV Scale for Teachers (CADS-T) total subscale score. CADS-T assesses symptoms of hyperactivity and inattention. The analysis of the primary efficacy variable showed a significant treatment difference in favor of Ritalin LA (p<0.0001). A statistically significant treatment effect for Ritalin LA relative to placebo was also found in all analyses of the secondary CADS efficacy variables, as well as in two post-hoc analyses for the ADHD diagnostic subtypes (combined type, inattentive type). The results of the primary and secondary efficacy analyses are summarized in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Adults with ADHD: Ritalin LA was evaluated in a randomized, double-blind, placebo-controlled, multicentre study (RIT124D2302) in the treatment of 725 adult patients (395 male and 330 female) diagnosed with ADHD according to DSM-IV ADHD criteria. The study was designed to: 1) Confirm the clinically effective and safe dose range of Ritalin LA for adults (18 to 60 years old) in a 9-week, double-blind, randomized, placebo-controlled, parallel group period (Period 1) consisting of a 3-week titration stage followed by a 6-week fixed dose stage (40, 60, 80 mg/day or placebo). Subsequently patients were re-titrated to their optimal dose of Ritalin LA (40, 60 or 80 mg/day) over a 5 week period (Period 2).
2) Evaluate the maintenance of effect of Ritalin LA in adults with ADHD in a 6-month, double-blind, randomized, withdrawal study (period 3).
Efficacy was assessed using the DSM-IV ADHD rating scale (DSM-IV ADHD RS) for symptomatic control and Sheehan Disability Score (SDS) for functional improvement as change in respective total scores from baseline to the end of the first period. All dose levels of Ritalin LA showed significantly greater symptom control (p<0.0001 for all dose levels) compared to placebo as measured by a reduction in DSM-IV ADHD RS total score. All doses of Ritalin LA showed significantly greater functional improvement (p=0.0003 at 40 mg, p=0.0176 at 60 mg, p<0.0001 at 80 mg) compared to placebo as measured by reduction in SDS total score (see Table 2).
Significant clinical efficacy was demonstrated at all three Ritalin LA dose levels using physician-rated scales [Clinical Global Impression-Improvement (CGI-I) and Clinical Global Improvement-Severity (CGI-S)], self-rated scales [Adult Self-Rating Scale (ASRS)] and observer-rated scales [Conners' Adult ADHD Rating Scale Observer Short Version (CAARS O: S)]. The results were consistently in favor of Ritalin LA over placebo across all assessments in period 1. (See Table 2.)
Click on icon to see table/diagram/image
Maintenance of effect of Ritalin LA was evaluated by measuring the percentage of treatment failure in Ritalin LA compared to the placebo group at the end of a 6-month maintenance period (see Table 3). Once the Ritalin LA dose was optimized in Period 2, approximately 79% of patients continued to maintain disease control for a period of at least 6 months (p <0.0001 vs. placebo). An odds ratio of 0.3 suggested that patients treated with placebo had a 3 times higher chance of becoming a treatment failure compared to Ritalin LA. (See Table 3.)
Click on icon to see table/diagram/image
Patients who entered Period 3 had completed a total of between 5-14 weeks of Ritalin LA treatment in Periods 1 and 2. Patients then assigned to placebo in Period 3 did not experience increased signs of withdrawal and rebound compared to patients who continued on Ritalin LA treatment.
The study performed in adults did not suggest any difference in efficacy or safety amongst gender subgroups (see DOSAGE & ADMINISTRATION).
The long-term efficacy and safety of Ritalin LA in adult patients was further evaluated in a 26-week open-label extension study of Ritalin LA in 298 adult patients with ADHD (RIT124D2302E1). Combining all patients in both studies, a total of 354 patients continuously received Ritalin LA for >6 months and 136 patients for >12 months.
The safety profile of Ritalin LA did not change with the longer duration of treatment of adult ADHD patients. The safety profile seen in study RIT124D2302E1 was similar to that observed in study RIT124D2302. No unexpected serious adverse events or adverse events were observed in this extension study and the commonly observed adverse events were expected and driven by the pharmacologic activity.
Furthermore, Ritalin LA treatment during the study consistently demonstrated clinical efficacy when using self-rated scales (SDS) and physician-rated scales (i.e. DSM-IV ADHD RS, CGI-I, and CGI-S). The results were consistently in favor of Ritalin LA treatment across all assessments. Patients continued to show symptomatic improvement and a reduction in functional impairment throughout the study as shown by the mean change in DSM-IV ADHD total score by -7.2 points and the mean change in SDS total score by -4.8 points when assessed against the extension baseline.
Pharmacokinetics: Absorption: Tablets: After oral administration the active substance (methylphenidate hydrochloride) is rapidly and almost completely absorbed. Owing to extensive first-pass metabolism, the absolute bioavailability was 22±8% for the d-enantiomer and 5±3% for the l-enantiomer. Ingestion with food has no relevant effect on the rate of absorption. Peak plasma concentrations of about 40 nmol/L (11 ng/mL) are reached on average 1 to 2 hours after administration. Peak plasma concentrations vary markedly between patients. The area under the concentration-time curve (AUC), and the peak plasma concentration (C
max) are proportional to the dose.
LA capsules: Following oral administration of Ritalin LA (modified-release capsules) to children diagnosed with ADHD and adults, methylphenidate is rapidly absorbed and produces a bi-modal plasma concentration-time profile (i.e. two distinct peaks approximately four hours apart). The relative bioavailability of Ritalin LA given once daily is comparable to the same total dose of Ritalin or methylphenidate tablets given twice a day in children and in adults.
The fluctuations between peak and trough plasma methylphenidate concentrations are smaller for Ritalin LA given once a day compared to Ritalin tablets given twice a day.
Ritalin LA may be administered with or without food. There were no differences in the bioavailability of Ritalin LA when administered with either a high-fat breakfast or apple sauce, compared to administration in the fasting condition. There is no evidence of dose dumping in the presence or absence of food.
For patients unable to swallow the capsule, the contents may be sprinkled on soft food such as apple sauce and administered (see DOSAGE & ADMINISTRATION).
Distribution: In blood, methylphenidate and its metabolites are distributed between plasma (57%) and erythrocytes (43%). Binding to plasma proteins is low (10 to 33%). The volume of distribution was 2.65±1.11 L/kg for d-MPH and 1.80±0.91 L/kg for l-MPH.
Methylphenidate excretion into breast milk has been noted in two case reports, where the calculated relative infant dose was ≤0.2% of the weight-adjusted maternal dose. Adverse events were not noted in either infants (6 and 11 months of age).
Biotransformation/metabolism: Biotransformation of methylphenidate by the carboxylesterase CES1A1 is rapid and extensive. Peak plasma concentrations of the main, deesterified, metabolite - alpha-phenyl-2-piperidine acetic-acid (ritalinic acid) - are attained about 2 hours after administration and are 30 to 50 times higher than those of the unchanged substance. The elimination half-life of alpha-phenyl-2-piperidine acetic acid is about twice that of methylphenidate, and its mean systemic clearance is 0.17 L/h/kg. Only small amounts of hydroxylated metabolites (e.g. hydroxymethylphenidate and hydroxyritalinic acid) are detectable. Therapeutic activity seems to be principally due to the parent compound.
Elimination: Methylphenidate is eliminated from the plasma with a mean elimination half-life of 2 hours. The systemic clearance is 0.40±0.12 L/h/kg for d-MPH and 0.73±0.28 L/h/kg for l-MPH. After oral administration, 78 to 97% of the dose is excreted in urine and 1 to 3% in feces in the form of metabolites within 48 to 96 hours. Only small quantities (<1%) of unchanged methylphenidate appear in the urine. Most of the dose is excreted in urine as alpha-phenyl-2-piperidine acetic acid (60 to 86%).
Special populations: Effect of age: There are no apparent differences in the pharmacokinetics of methylphenidate between hyperactive children (6-13 years) and healthy adult volunteers.
Patients with renal impairment: Elimination data from patients with normal renal function suggest that renal excretion of unchanged methylphenidate would hardly be diminished in the presence of impaired renal function. However, renal excretion of the metabolite alpha-phenyl-2-piperidine acetic acid may be reduced.
Toxicology: Non-Clinical Safety Data: Reproductive toxicity: See USE IN PREGNANCY & LACTATION.
Fertility: Methylphenidate did not impair fertility in male or female mice that were fed diets containing the drug in an 18-week continuous breeding study. The study was conducted over two generations of mice continuously receiving methylphenidate doses of up to 160 mg/kg/day (about 90-fold higher than the MRHD on a mg/kg basis).
Carcinogenicity: In a lifetime carcinogenicity study carried out in B6C3F1 mice, methylphenidate caused an increase in hepatocellular adenomas (a benign tumor) and, in males only, an increase in hepatoblastomas (a malignant tumor) at daily doses of approximately 60 mg/kg/day about 35-fold-higher than the maximum recommended human dose (MRHD) on a mg/kg basis. Hepatoblastoma is a relatively rare rodent malignant tumor type. There was no overall increase in the number of malignant hepatic tumors. The mouse strain used is particularly sensitive to the development of hepatic tumors. It is thought that hepatoblastomas might be due to non-genotoxic mechanisms such as an increase in hepatic cell proliferation. This is consistent with the increase in liver weights observed in this mouse carcinogenicity study.
Methylphenidate did not cause any increase in tumors in a lifetime carcinogenicity study carried out in F344 rats; the highest dose used was approximately 45 mg/kg/day (about 26-fold higher than the MRHD on a mg/kg basis).
Genotoxicity: With methylphenidate, sister chromatid exchange and chromosome aberrations were elevated in one
in vitro study in Chinese Hamster Ovary (CHO) cells. However, no genotoxicity effects were seen in several other assays, including no mutagenic effects in three
in vitro tests (Ames reverse mutation test, mouse lymphoma forward mutation test, human lymphocyte chromosome aberration test) and no evidence of clastogenic or aneugenic effects in two
in vivo mouse bone marrow micronucleus tests, at doses up to 250 mg/kg. B6C3F1 mice from the same strain that showed liver tumors in the cancer bioassay were used in one of these studies.
Additionally, there was no genotoxic potential as assessed by measuring cII mutations in the liver and micronuclei in peripheral reticulocytes in the Big Blue mouse, micronuclei in peripheral blood reticulocytes, HPRT mutations and chromosomal aberrations in peripheral blood lymphocytes of rhesus monkeys, Pig-A locus mutations in adolescent rats, micronucleated reticulocyte frequencies in blood and DNA damage in blood, brain, and liver cells of adult male rats treated for 28 consecutive days, and by measuring micronuclei in mouse peripheral blood erythrocytes.
Juvenile toxicity: In a conventional study conducted in young rats, methylphenidate was administered orally at doses of up to 100 mg/kg/day for 9 weeks, starting early in the postnatal period (postnatal day 7) and continuing through sexual maturity (postnatal week 10). When the animals were tested as adults (postnatal weeks 13-14), decreased spontaneous locomotor activity was observed in males and females previously treated with 50 mg/kg/day or greater, and a deficit in the acquisition of a specific learning task was seen in females exposed to the highest dose of 100 mg/kg/day (about 58-fold higher than the MRHD on a mg/kg basis). The clinical relevance of these findings is unknown.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image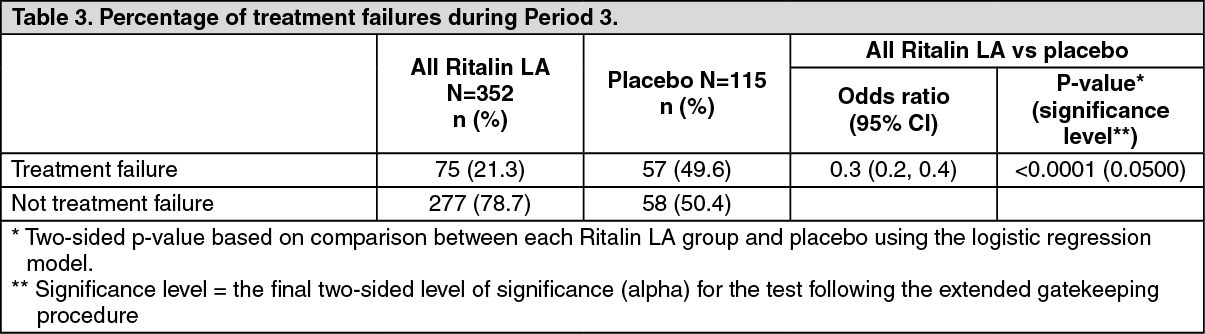 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image



 Sign Out
Sign Out
